| LaSpHUB | 
|
| Home | LaspMol | Mol on Surface | MolonSurf GPTs| |
| LaSpHUB |
|
| Home | LaspMol | Mol on Surface | MolonSurf GPTs| |
|
Home
LASP History Mannual Examples NN Library LASP Download LASP Methodology LASP Applications LASPview LASP Tutorial LiuGroup at Fudan Job position Available |
LASP Applications Highlight
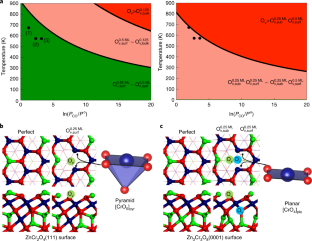
AbstractMetal oxide alloys (for example AxByOz) exhibit dramatically different catalytic properties in response to small changes in composition (the A:B ratio). Here, we show that for the ternary zinc–chromium oxide (ZnCrO) catalysts the activity and selectivity during syngas (CO/H2) conversion strongly depend on the Zn:Cr ratio. By using a global neural network potential, stochastic surface walking global optimization and first principles validation, we constructed a thermodynamics phase diagram for Zn–Cr–O that reveals the presence of a small stable composition island, that is, Zn:Cr:O = 6:6:16 to 3:8:16, where the oxide alloy crystallizes into a spinel phase. By changing the Zn:Cr ratio from 1:2 to 1:1, the ability to form oxygen vacancies increases appreciably and extends from the surface to the subsurface, in agreement with previous experiments. This leads to the critical presence of a four-coordinated planar Cr2+ cation that markedly affects the syngas conversion activity and selectivity to methanol, as further proved by microkinetics simulations
AbstractGlucose pyrolysis, a model system in biomass utilization, is renowned for its great complexity, deep in reaction network hierarchy and rich in reaction patterns. The selectivity in glucose pyrolysis, e.g., the high yield of 5-hydroxymethylfurfural (HMF), a value-added platform product, remains an intriguing puzzle even after 60 years of experimental study. Here we resolve the whole reaction network of glucose pyrolysis using a global-to-global technique for reaction pathway sampling. This is achieved by establishing the first organic chemistry reaction database via stochastic surface walking (SSW) global optimization, building the global neural network (G-NN) potential via machine learning and extensively exploring the reaction network of glucose pyrolysis. In total, 6407 elementary reactions, screened out from more than 150 000 reaction pairs in glucose pyrolysis, are collected in our reaction database. The established reaction network from SSW-NN, further validated by first-principles calculations, reveals that for glucose to HMF, the lowest energy reaction pathway involves fructose and 3-deoxyglucos-2-ene (3-DGE) as key intermediates and a site-selective reaction type, retro-Michael-addition, for three consecutive dehydration steps. The overall barrier is determined to be 1.91 eV, being at least 0.19 eV lower than all previously proposed mechanisms, which assumes direct β-H elimination dehydration. The lowest pathways to the other two major products, furfural (FF) and hydroxyacetaldehyde (HAA), are also discovered with a similar barrier 1.95 eV, which exhibit a competing nature by sharing the same key intermediate, 3-ketohexose. Since chemical reactions occurring in fast glucose pyrolysis are generally present in biomass chemistry, containing essentially all reaction patterns of C–H–O elements, the methodology designed and the results presented would help to advance reaction design and mechanistic modeling in renewable fuels from biomass.
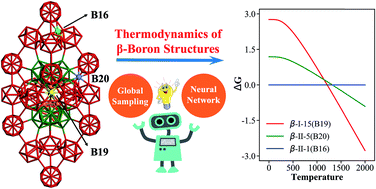
AbstractBoron crystals, despite their simple composition, must rank top for complexity: even the atomic structure of the ground state of β-B remains uncertain after 60 years’ study. This makes it difficult to understand the many exotic photoelectric properties of boron. The presence of self-doping atoms in the crystal interstitial sites forms an astronomical configurational space, making the determination of the real configuration virtually impossible using current techniques. Here, by combining machine learning with the latest stochastic surface walking (SSW) global optimization, we explore for the first time the potential energy surface of β-B, revealing 15 293 distinct configurations out of the 2 × 105 minima visited, and reveal the key rules governing the filling of the interstitial sites. This advance is only allowed by the construction of an accurate and efficient neural network (NN) potential using a new series of structural descriptors that can sensitively discriminate the complex boron bonding environment. We show that, in contrast to the conventional views on the numerous energy-degenerate configurations, only 40 minima of β-B are identified to be within 7 meV per atom in energy above the global minimum of β-B, most of them having been discovered for the first time. These low energy structures are classified into three types of skeletons and six patterns of doping configurations, with a clear preference for a few characteristic interstitial sites. The observed β-B and its properties are influenced strongly by a particular doping site, the B19 site that neighbors the B18 site, which has an exceptionally large vibrational entropy. The configuration with this B19 occupancy, which ranks only 15th at 0 K, turns out to be dominant at high temperatures. Our results highlight the novel SSW-NN architecture as the leading problem solver for complex material phenomena, which would then expedite substantially the building of a material genome database.
AbstractAmorphous structures are often good catalysts for their large varieties of exposed surface sites, but the characterization of the catalytic sites has long been a great challenge for both theory and experiment. One such interesting example is the recently synthesized “black TiO2” via the hydrogenation of TiO2 nanoparticles, which has an amorphous shell and crystalline core, and exhibits high hydrogen evolution reaction (HER) activity in the visible-light photocatalytic water splitting. Here, we utilize our recently developed theoretical tool, namely, the stochastic surface walking global optimization with neural network potential followed by first-principles validation, to quantitatively determine the thermodynamic phase diagram of TiO2 in contact with H2 at different temperatures and pressures. Among a number of anatase surfaces, we show that, only on a ridged anatase (112) surface, a local high H coverage, 0.69 ML, can be gradually built up accompanied by the surface amorphization. This high H coverage not only renders the black color of the amorphous TiO2 but also provides an unprecedented low-energy reaction channel for the HER: a transient Ti–H hydride becomes likely to form on exposed Ti atoms, and its reaction with neighboring OH has much lower barrier, i.e., 0.6 eV, compared to the traditional H coupling channel via two surface OH groups (barrier >1.6 eV). Our results not only provide deep insights into the surface species and reactions that are unique to amorphous materials, but also demonstrate that the global sampling with neural network potential holds a great promise for solving complex structures under realistic reactions.
AbstractSeeking for active MnOx material as artificial water splitting catalyst has been a long history since the discovery of PSII system in nature. To date, the highest activity MnOx catalyst reported for oxygen evolution reaction (OER) does however not belong to common MnO2 polymorphs (α-, β-, δ-MnO2), but rather to nascent δ-MnO2 layer produced in situ from spinel under electrochemical conditions with unknown active site structure. Here with the stochastic surface walking (SSW) pathway sampling method, we for the first time resolve the atomic-level mechanism of spinel-to-layer Mn3O4 solid phase transition in aqueous electrolyte. We show that a transient H0.5MnO2 phase is the precursor of transition that forms at high voltage (>1 V), and it undergoes the solid-to-solid phase transition to produce a δ-MnO2 layer, which is accompanied by Mn dissolution, dislocation, layer-breaking, and insertion of water/cations between layers. This leads to the generation of a variety of possible defective structures. We demonstrate using first-principles calculations that a special edge site with neighboring Mn vacancy provides the best OER activity with an overpotential of 0.59 V, 0.19 V lower than that of pristine MnO2. The high activity of such Mn sites are attributed to its special local structure: pseudocubane with one corner missing. The presence of the Mn vacancy near the active site enhances the adsorption of OH intermediate in OER. This defective cubane structure shares the common geometrical and electronic features found in the PSII system.
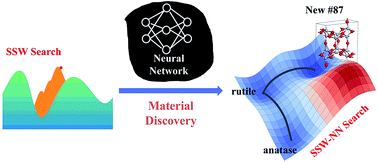
AbstractWhile the underlying potential energy surface (PES) determines the structure and other properties of a material, it has been frustrating to predict new materials from theory even with the advent of supercomputing facilities. The accuracy of the PES and the efficiency of PES sampling are two major bottlenecks, not least because of the great complexity of the material PES. This work introduces a “Global-to-Global” approach for material discovery by combining for the first time a global optimization method with neural network (NN) techniques. The novel global optimization method, named the stochastic surface walking (SSW) method, is carried out massively in parallel for generating a global training data set, the fitting of which by the atom-centered NN produces a multi-dimensional global PES; the subsequent SSW exploration of large systems with the analytical NN PES can provide key information on the thermodynamics and kinetics stability of unknown phases identified from global PESs. We describe in detail the current implementation of the SSW-NN method with particular focuses on the size of the global data set and the simultaneous energy/force/stress NN training procedure. An important functional material, TiO2, is utilized as an example to demonstrate the automated global data set generation, the improved NN training procedure and the application in material discovery. Two new TiO2 porous crystal structures are identified, which have similar thermodynamics stability to the common TiO2 rutile phase and the kinetics stability for one of them is further proved from SSW pathway sampling. As a general tool for material simulation, the SSW-NN method provides an efficient and predictive platform for large-scale computational material screening.
AbstractUnder mild static compression (15 GPa), graphite preferentially turns into hexagonal diamond, not cubic diamond, the selectivity of which is against thermodynamics. Here we, via novel potential energy surface global exploration, report seven types low energy intermediate structures at the atomic level that are key to the kinetics of graphite to diamond solid phase transition. On the basis of quantitative kinetics data, we show that hexagonal diamond has a facile initial nucleation mechanism inside graphite matrix and faster propagation kinetics owing to the presence of three coherent graphite/hexagonal diamond interfaces, forming coherent nuclei in graphite matrix. By contrast, for the lack of coherent nucleus core, the growth of cubic diamond is at least 40 times slower and its growth is inevitably mixing with that of hexagonal diamond.
AbstractAs a model system of 2-D oxide material, layered δ-MnO2 has important applications in Li ion battery systems. δ-MnO2 is also widely utilized as a precursor to synthesize other stable structure variants in the MnO2 family, such as α-, β-, R-, and γ-phases, which are 3-D interlinked structures with different tunnels. By utilizing the stochastic surface walking (SSW) pathway sampling method, we here for the first time resolve the atomistic mechanism and the kinetics of the layer-to-tunnel transition of MnO2, that is, from δ-MnO2 to the α-, β-, and R-phases. The SSW sampling determines the lowest-energy pathway from thousands of likely pathways that connects different phases. The reaction barriers of layer-to-tunnel phase transitions are found to be low, being 0.2–0.3 eV per formula unit, which suggests a complex competing reaction network toward different tunnel phases. All the transitions initiate via a common shearing and buckling movement of the MnO2 layer that leads to the breaking of the Mn–O framework and the formation of Mn3+ at the transition state. Important hints are thus gleaned from these lowest-energy pathways: (i) the large pore size product is unfavorable for the entropic reason; (ii) cations are effective dopants to control the kinetics and selectivity in layer-to-tunnel transitions, which in general lowers the phase transition barrier and facilitates the creation of larger tunnel size; (iii) the phase transition not only changes the electronic structure but also induces the macroscopic morphology changes due to the interfacial strain.
AbstractThe solid phase transition of TiO2, in particular anatase to rutile, has been extensively studied in the past 30 years. To seek the nucleation site at the beginning of phase transition is highly challenging, which asks for new theoretical techniques with high spatial and temporal resolution. This work reports the first evidence on the atomic structure of the nucleation sites in the TiO2 anatase-to-rutile phase transition. Novel automated theoretical methods, namely stochastic surface walking based pathway sampling methods, are utilized to resolve the lowest energy pathways at the initial stage of phase transition. We show that among common anatase surfaces, only the (112) ridged surface provides the nucleation site for phase transition, which can lead to the formation of both TiO2-II and brookite thin slabs. The TiO2-II phase is kinetically preferred product; the propagation into the subsurface is still hindered by high barriers that is the origin for the slow kinetics of nuclei formation. The rutile nuclei are thus not rutile phase but nascent metastable TiO2-II phase in an anatase matrix. The phase transition kinetics is found to be sensitive to the compressive strain and the crystallographic directions. The results rationalize the size and morphology dependence of the anisotropic phase transition kinetics of anatase particles and could facilitate the rational design of material via controlled solid phase transition.
AbstractThe solid-phase transitions of zirconia are important phenomena for many industrial applications. Because of the lack of tools for resolving the atom displacement pattern, the transition kinetics has been disputed for over 60 years. Here, first-principles-based stochastic surface walking (SSW) pathway sampling is utilized for resolving the mechanism of ZrO2 tetragonal-to-monoclinic solid-phase transition. Two types of lattice and atom correspondence allowed in phase transition are determined for the first time from energy criterion, which are originated from two nearly energy-degenerate lowest-transition pathways and one stress-induced ferroelastic transition channel of tetragonal phase. An orthorhombic crystal phase (Pbc2/1) is discovered to be a trapping state at low temperatures in phase transition, the presence of which does not create new orientation relation but deters transformation toughening significantly. This new finding may facilitate the design of new functional oxide materials in ceramic industry.
|
版权所有:上海鞍面智能科技有限公司 2018--2028 ver 1.00 沪ICP备18016621号-1 |